
Fig. 1: We are working on the molecular mechanisms of ageing, with a focus on the multi-faceted roles of NAD+ in healthy longevity.

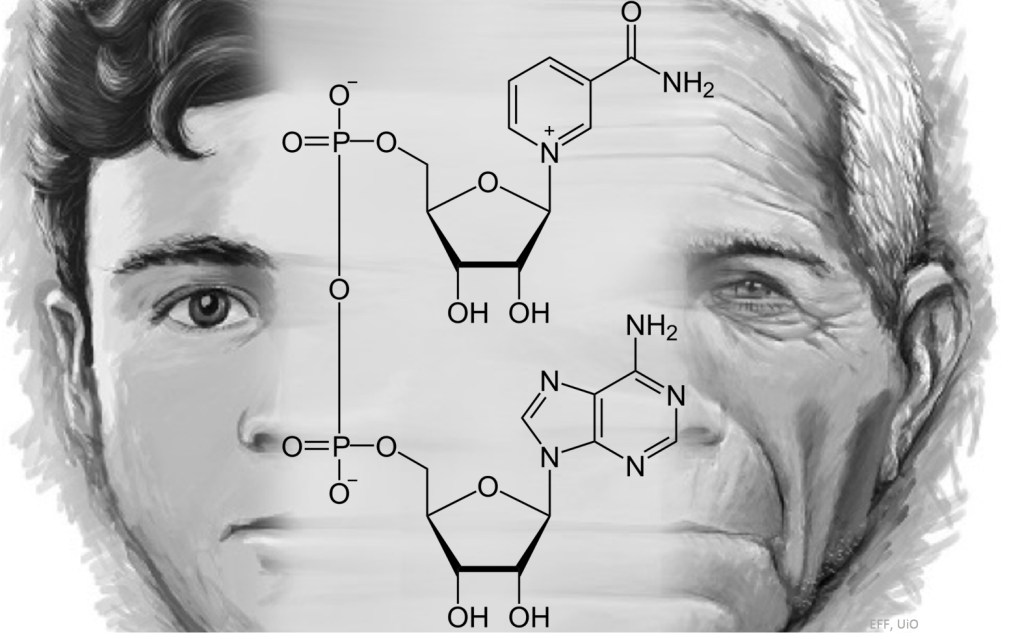
Fig. 2: The NAD+ synthetic pathways and the NAD+-consuming enzymes. Image: Fang EF et al., Trends Molecular Medicine, 2017.
(A) NAD+ is produced via three major pathways in mammals. The first is the de novo biosynthesis from tryptophan (Trp), also called the kynurenine pathway. Trp enters the cell via the transporters SLC7A5 and SLC36A4. Within the cell, Trp is converted to formylkynurenine (FK), which is further converted to kynurenine. Kynurenine can be converted to kynurenic acid (via kynurenine aminotransferases/KATs) and finally quinaldic acid. Additionally, kynurenine can be converted to 3-hydroxykynurenine (3-HK) by kynurenine 3-monooxygenase (KMO), and further to 3-hydroxyanthranilic acid (3-HAA) by tryptophan 2,3-dioxygenase (KYNU). The next step is performed by 3-hydroxyanthranilic acid oxygenase (3HAO) to produce α-amino-β-carboxymuconate-ϵ-semialdehyde (ACMS). Via a spontaneous reaction, ACMS converts to quinolinic acid, which further formulates to NAMN by quinolinate phosphoribosyltransferase (QPRT), and to nicotinic acid adenine dinucleotide (NAAD), and finally to NAD+. 3-HK, 3-HAA, and quinolinic acid are neurotoxic (denoted with red asterisk), whereas kynurenic acid and picolinic acid are neuroprotective (marked with green asterisk). The second pathway is the Preiss-Handler pathway during which nicotinic acid (NA) is used as an NAD+ precursor. NA enters the cell via SLC5A8 or SLC22A3 transporters. The Preiss-Handler pathway is initiated by the conversion of NA to NAMN by NA phosphoribosyl-transferase (NAPRT). NAMN, an intermediate in both kynurenine pathway and the Preiss-Handler pathway, is converted to form NAAD by NAM mononucleotide transferases (NMNATs). Lastly, NAAD is converted to NAD+ by NAD+ synthase (NADS). The third pathway is the salvage pathway with the cells generating NAD+ from nicotinamide riboside (NR) and recycling nicotinamide (NAM) back to NAD+ via nicotinamide mononucleotide (NMN). Extracellularly, NAD+ or NAM can be converted to NMN, which is in turn dephosphorylated to NR, possibly by CD73. NR is transported into the cell via an unknown nucleoside transport. Intracellularly, NR forms NMN via NRK1 or NRK2 in a tissue-specific manner. NMN is then converted to NAD+ by NMNATs. The enzyme NAM N-methyltransferase (NNMT) methylates NAM, using S-adenosyl methionine (SAM) as a methyl donor. This removes NAM from recycling and indirectly affects NAD+ levels. (B) The four major NAD+-consuming enzymes. From left: poly(ADP-ribose) polymerases (PARPs), especially PARP1 and PARP2, use NAD+ as a co-substrate to PARylate target proteins, generating NAM as a by-product. The deacetylation activity of sirtuin (SIRT)1, SIRT3, and SIRT6 depends on NAD+, generating NAM as a by-product, with NAM at high cellular levels inhibiting the activity of SIRTs. The NADases or cyclic ADP-ribose synthases (cADPRSs) CD38 and CD157 hydrolyze NAD+ to NAM, generating ADPR and cADPR; in addition, CD38 can degrade NMN to NAM, removing NMN from NAD+ synthesis. Sterile alpha and TIR motif-containing 1 (SARM1) was recently identified as a NADase, which cleaves NAD+ to NAM, cADPR, and ADPR. (C) The chemical structures of NAD+ and the NAD+ precursors. From left: nicotinic acid (NA), nicotinamide (NAM), nicotinamide riboside (NR), nicotinamide mononucleotide (NMN), and oxidized form of nicotinamide adenine dinucleotide (NAD+). Note that CD38, CD73, and CD157 are membrane proteins. For simplicity, we did not attach them to the membrane in this schematic model.

Fig. 3. Proposed mechanism of the occurrence of the earliest AD pathology in entorhinal cortex (EC) L-II: linkages of impaired mitophagy which leads to impaired mitochondrial homeostasis, in Abeta and Tau pathologies are presented. (A) Immunofluorescently labeled reelin-positive neuronal population in L-II of rat EC. (B-C) Schematic representation of the signal transduction cascade generated by reelin under normal physiological conditions (B) vs AD (C). (B) Under normal conditions, including in young individuals, reelin is highly expressed. Reelin binds to lipoprotein receptors, such as ApoER2 and VLDLR, inducing activation of Disabled 1 (Dab1), an adapter protein. Activated Dab1 induces activation of Src family kinases (SFKs) that potentiate tyrosine phosphorylation of Dab1, which in turn activates Phosphoinositide 3-kinase (PI3K) and subsequently protein kinase B (PKB). PKB activation inhibits the activity of GSK3β, thereby reducing p-Tau and promoting microtubule stability. PKB also activates mTOR-dependent processes which promote the outgrowth of dendrites and balances mitochondrial biogenesis and autophagy. Changes of mTOR activity affect the activities of ULK1, AMPK, sirtuins (SIRTs), FOXOs, and NAD+. In addition, ApoER2-reelin complex is coupled to NMDAR signaling through PSD95. Reelin-activated SFK phosphorylates NMDAR and potentiates NMDAR-Ca2+ influx. Influx of Ca2+ activates the transcriptional regulator, CREB, through which expression of genes important for synaptic plasticity and neurite growth are potentiated. CREB-regulated genes encode proteins important for learning and memory. Astrocyte- and neuron-derived ApoEs bind to ApoER2 and are constitutively internalized. Upon reelin signaling, ApoER2 also undergoes endocytosis. In the cases of ApoE2 and ApoE3, ApoER2 is efficiently recycled to the cell membrane; this is impaired in the case of ApoE4. In healthy young individuals, mitophagy effectively clears damaged mitochondria, ensuring a healthy mitochondrial pool in the high-energy demanding axon terminals; this enables normal neuronal function and neuronal plasticity. Mitophagy also eliminates intracellular iAβ1-42 and pathological Tau proteins. (C) Ageing is the primary driver of AD with multiple molecular mechanisms involved, including age-dependent reduction of reelin and impaired mitophagy (also autophagy). In prodromal AD, reduced reelin in the EC L-II neurons impairs the control of the ApoER2/VLDLR-Dab-1-PI3K-PKB-GSK3β axis, leading to pTau. Furthermore, iAβ1-42 is increased in the EC L-II neurons, possibly due to increased production (via ApoE4-dependent trapping, detailed below), and reduced clearance by impaired mitophagy/autophagy. iAβ1-42 may bind reelin and thereby reduce levels of signaling competent reelin, in turn impairing PKB mediated inhibition of GSK3β and thus increasing p-Tau. ApoE4 may accentuate this as it tends to get trapped in endosomes along with its lipoprotein receptors, likely mainly ApoER2. This in turn further reduces reelin-signaling, boosting the cascade leading to p-Tau. In concert, ApoE4, trapped in endosomes, increases transcription of APP and thus production of iAβ1-42, and thereby completes a vicious cycle, whose end product for the affected neurons are NFTs. Furthermore, reduced PKB-activity also inhibits mTOR activity, impacting on mitochondrial homoeostasis and autophagy. Moreover, ApoE4 sequesters ApoER2 in intracellular compartments and reduces the NMDAR phosphorylation in response to reelin in the postsynaptic neuron, leading to impaired neural plasticity. In line with the age onset of AD, age-dependent mitophagy impairment causes accumulation of damaged mitochondria, which further exacerbates AD pathology, including shortage of energy supply, inflammation, oligomerization of iAβ1-42 and pathological Tau proteins, finally leading to impaired LTP and neuronal plasticity, and neuronal loss. Individuals carrying ApoE4 may have exacerbated mitophagy impairment since ApoE4 inhibits TFEB-dependent regulation of autophagy- and lysosome-related genes. Dashed lines and faint phosphorylation symbols indicate blunted signaling capacity. Abbreviations: Aβ, amyloid-β; AD, Alzheimer’s disease; AMPK, 5′ AMP-activated protein kinase; ApoE, apolipoprotein E; ApoER2, ApoE receptor 2; Ca2+, calcium ion; CREB, cAMP response element-binding protein; Dab1, disabled 1; EC, entorhinal cortex; FOXOs, Forkhead box O (FOXO) transcription factors; GSK3β, glycogen synthase kinase 3β; LTP, Long-term potentiation; NMDAR, N-methyl-D-aspartate receptor; PKB, protein kinase B; SIRTs, the NAD+-dependent deacetylates sirtuins; SFKs, SRC family tyrosine kinases; ULK1, unc-51 like autophagy activating kinase 1; VLDLR, very-low-density lipoprotein receptor; low-density lipoprotein receptor-related protein 1 (LRP1). Dashed arrows indicate impaired induction/activation.
Figure: Asgeir Kobro-Flatmoen…Evandro F. Fang, Ageing Research Reviews 2021
https://doi.org/10.1016/j.arr.2021.101307

Fig. 4. Impaired mitochondrial clearance in AD neurons. (A) In a healthy neuron, mitochondrial homeostasis is maintained through different pathways, such as mitophagy, UPSmt, and MDVs. Detailed molecular mechanisms of these pathways are visualized in Fig. 2. Neuronal mitophagy likely happens predominantly in the soma, with existence of in-site mitophagic degradation in the axon, including in the axon terminals. Mitochondria are transported from the soma to distal parts of neurons or to the sites of high energy demand along microtubule tracks using the motor protein complex kinesin (anterograde direction) or dynein (retrograde direction). In healthy neurons, damaged mitochondria in the distal regions of the axon can either be retrogradely transferred to the soma for mitophagic degradation or can be on-site degraded via mitophagy by lysosomes anterogradely transferred from the soma. (B) Impaired mitophagy and enhanced pathologies in the AD neurons. In AD neurons, increased accumulation of iAβ1-42 and p-Tau leads to abnormal trafficking in both directions. Aβ toxic oligomers and p-Tau are likely involved in microtubule destabilization, impairing retrograde transportation of damaged mitochondria from distal regions back to the soma for degradation, and may block anterograde transport of kinesin-tagged lysosomes to axons for on-site mitophagic degradation, resulting in impaired local degradation capacity. Impaired mitophagy is likely to exacerbate AD pathology by reducing the neurons’ capacity for degradation of toxic proteins, including iAβ1-42 and p-Tau. While evidence exists for impaired mitophagy and compromised UPSmt in AD, the status of MDV in AD is not known.
Figure: Asgeir Kobro-Flatmoen…Evandro F. Fang, Ageing Research Reviews 2021
https://doi.org/10.1016/j.arr.2021.101307

Fig. 5. The use of artificial intelligence to propel screening of novel molecules which can increase healthy longevity (from the 4th century AD to the 21st century). This is the central roundel of a 4th-century AD mosaic floor from a villa at Hinton St. Mary, Dorset. It is one of the most important early Christian remains from the Roman Empire. The roundel is probably the earliest known mosaic picture of Christ. At either side are pomegranates, signalling immortality. We have reported that a small compound Urolithin A (UA), affluent in pomegranate, inhibits memory loss in different animal models of Alzheimer´s disease (Fang EF et al., Nature Neuroscience 2019). Very recently, we have used an AI approach and identified two lead compounds showing potential anti-AD capacity.
Image: left, Evandro Fang taken from The British Museum; right, a book chapter on AI in medical use with the image generated by Alice Rui-xue Ai from the Fang group.

Fig. 6. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. An example of a recent success we made: to use AI to propel drug development against Alzheimer’s disease. The paper.
Research topics of the Fang group
We are fascinated with the molecular mechanisms of human ageing. We work on several ageing theories, including DNA damage, mitochondrial dysfunction, and stem cell exhaustion. The final goal is to apply our findings to the development of effective interventional strategies for human aging and age-related diseases. There are three major research topics in the laboratory:
1. NAD+ in ageing (Figures 1-2)
The coenzyme NAD+ is critical in cellular bioenergetics and adaptive stress responses. Its depletion has emerged as a fundamental feature of ageing that may lead to a wide range of chronic diseases. Maintenance of NAD+ levels is important for cells with high energy demand and for proficient neuronal function. NAD+ depletion is detected in major neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases, cardiovascular disease, muscle atrophy, and accelerated aging diseases. Emerging evidence suggests that NAD+ decreases in various tissues during aging, and that physiological and pharmacological interventions bolstering cellular NAD+ levels might retard aspects of ageing and forestall some age-related diseases. We are interested in the networks between NAD+ and the 10 hallmarks of ageing. We work on the molecular mechanisms of NAD+ metabolism and consumption, and are exploring its clinical applications.
2. Defective mitophagy as a new contributor of Alzheimer´s disease (Figures 3-4)
2.1. Mechanisms of mitophagy.
Mitochondria are the powerhouse of the cell. There is an age-dependent deterioration in the quality of mitochondria. Mitochondrial dysfunction contributes to normal aging and to a wide spectrum of age-related diseases. Thus, it is important to maintain a healthy mitochondrial population, which is tightly regulated by proteolysis and mitophagy. Mitophagy is a specialized form of autophagy that regulates the turnover of damaged and dysfunctional mitochondria. Mechanistic studies on mitophagy across species highlight a sophisticated and integrated cellular network that regulates the degradation of mitochondria. We work on the discovery of new mitophagy proteins and how age affects mitophagy machinery.
Image: Kerr JS et al. Fang EF, Trends in Neuroscience 2017
2.2 Mechanisms of defective mitophagy in AD
Alzheimer’s disease (AD) affects 44 million people worldwide and causes formidable economic challenges. Continued failures in clinical anti-AD drug development suggest successful treatments may be found by investigating other molecular mechanisms in AD. AD neurons experience mitochondrial dysfunction and a bioenergetic deficit that occurs early and leads to the disease- defining Ab and Tau pathologies. Emerging findings suggest that the autophagy/lysosome pathway that removes damaged mitochondria (mitophagy) is also compromised in AD, resulting in the accumulation of dysfunctional mitochondria. Results in animal and cellular models of AD and in patients with sporadic late-onset AD suggest that impaired mitophagy contributes to AD; however, molecular mechanisms remain largely unexplored 4. We are extremely interested in the molecular mechanisms of defective mitophagy in AD and how mitophagy deficiency contributes to AD progression… Strategies directed at maintaining a healthy mitophagy level in AD might have beneficial effects. We are now using AD human iPSCs and AD animal models (mice and C. elegans) in combination with cutting-edge proteomics, microarray, imaging and biochemistry techniques to approach these questions. Our final goal is to unveil AD etiology and discover novel AD drug candidates, which may be more likely to succeed in the clinic.
Image: Kerr JS et al. Fang EF, Trends in Neuroscience 2017
3. The application of artificial intelligence in screening small compounds for Alzheimer´s disease and healthy longevity (Figure 5)
In view of the importance of mitophagy in neuroprotection and healthy longevity, we aim to screen robust but non-toxic mitophagy inducers. We collaborate with the leading artificial intelligence (AI) company Aladdin to propel the speed of drug development.





